恒水过滤带您详细了解除菌滤芯验证一、关于除菌过滤验证
除菌过滤验证包含除菌过滤器本身的性能确认和过滤工艺验证两部分。除菌过滤器性能确认和过滤工艺验证,两者很难互相替代,应独立完成。
除菌过滤器本身的性能确认一般由过滤器生产商完成;除菌过滤工艺验证可以由过滤器的使用者或委托试验检测机构(例如:过滤器的生产者或第三方实验室)完成,但过滤器使用者应最终保证实际生产过程中操作参数和允许的极值在验证时已被覆盖,并有相应证明文件。
小结:
1. 不同过滤器生产商的验证文件一般是不能相互替代的,同一生产商的同一材质的除菌过滤验证文件往往也不能直接互换,除非有合理的声明或文件支持。 二、细菌截留试验
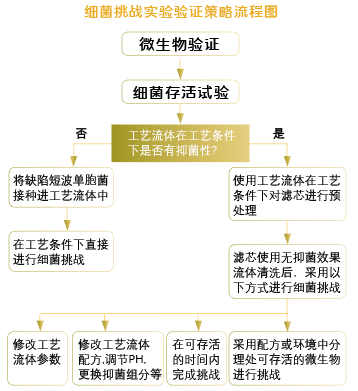
细菌截留试验的研究目的是模拟实际生产过滤工艺中的最差条件(过滤温度相对越高,过滤时间越长,批量越大,压差越高越是最差条件),过滤含有一定量挑战微生物的产品溶液或者产品替代溶液,以确认除菌过滤器的微生物截留能力。在进行细菌截留试验测试前,活度实验(生存性实验),确认挑战微生物于工艺条件下在药品中的存活情况,以确定合理的细菌挑战方法。
小结: 1. 缺陷型假单胞菌是除菌过滤验证中细菌截留试验的标准挑战微生物。
2. 微生物截留试验通常包括三个批次,其中至少应有一个批次为低起泡点(低规格)滤膜。
3. 低泡点要求:接近标准泡点值10%范围内。
4. 如果在验证中没有使用低起泡点滤膜,那么在实际生产中所使用的标准溶液滤膜/芯起泡点值,必须高于验证试验中实际使用的滤膜的最小起泡点值。
5. 设置一个0.45μm平行对照。
6. 应尽可能将挑战微生物直接接种在药品中进行细菌挑战。
7. 如果使用替代溶液进行试验,需要提供合理的数据和解释。
8. 对于同一种产品,即具有相同组分而不同浓度的产品,可以用挑战极限浓度的方法进行验证。
三、可提取物和浸出物试验
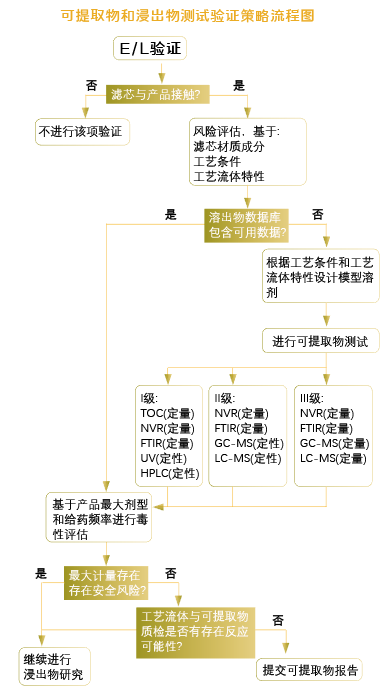
可提取物:是指通过使用模拟的或更加苛刻的作用力(例如,溶剂、温度或时间)可能会从接触表面萃取的化学成分。在模拟溶剂和“最差”条件下确定。(模型溶剂的选择、灭菌条件参数、过滤最高温度、接触最长时间等最差条件。)
小结: 1. 应先获得最差条件下的可提取物数据,将其用于药品的安全性评估。可提取物反映了浸出物的最大可能,无论是否要做浸出物试验,可提取物的测试和评估都非常重要。
2. 用于测试的模型溶剂应能够模拟实际的药品处方,同时与过滤器不应有化学兼容性方面的问题。通常应具有与产品相同或相似的理化性质,如pH值、极性或离子强度等。
3. 使用最长过滤时间、最高过滤温度、最多次蒸汽灭菌循环、增加伽玛辐射的次数和剂量都可能会增加可提取物水平。
4. 可提取物试验应使用灭菌后的滤器来完成。用于试验的过滤器尽量不进行预冲洗。
5. 可提取物和浸出物的检测需要采用定性和定量结合的方法。
6. 在完成可提取物或者浸出物试验后,应针对过滤器可提取物或浸出物的种类和含量,结合药品最终剂型中的浓度、剂量大小、给药时间、给药途径等对结果进行安全性评估,以评估可提取物和浸出物是否存在安全性风险。
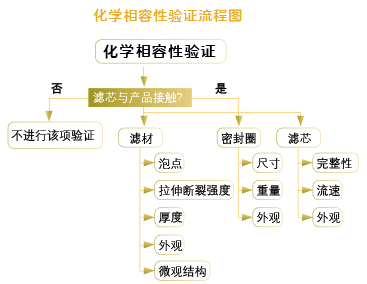
四、化学相容性试验化学相容性试验的研究目的:用来评估在特定工艺条件下,待过滤介质对过滤装置的化学影响。
1. 化学相容性试验应涵盖整个过滤装置,不只是滤膜,除了滤膜,其外壳骨架、密封圈、支撑导流层均需要考虑。
2. 应考虑率最差工艺条件,过滤流速、接触时间和温度等。
3. 化学相容性试验检测项目一般包括:过滤器接触待过滤介质前后的目视检查;过滤过程中流速变化;滤膜重量/厚度的变化;过滤前后起泡点等完整性测试数值的变化;滤膜拉伸强度的变化;滤膜电镜扫描确认等。
五、吸附试验
2. 过滤器中吸附性的材料包括滤膜、外壳和支撑性材料。流速、过滤时间、待过滤介质浓度、防腐剂浓度、温度和pH值等因素都可能影响吸附效果。
六、基于产品完整性试验 |
| 法规 |
欧盟 EME/APICS |
中国 NMPA |
美国 FDA |
WHO CGMP |
|
灭菌前 |
--- |
可以测试 |
可以测试 |
可以测试 |
|
灭菌后,使用前 |
应该测试 |
可以测试 |
可以测试 |
可以测试 |
|
使用后 |
应该测试 |
应该测试 |
应该测试 |
应该测试 |
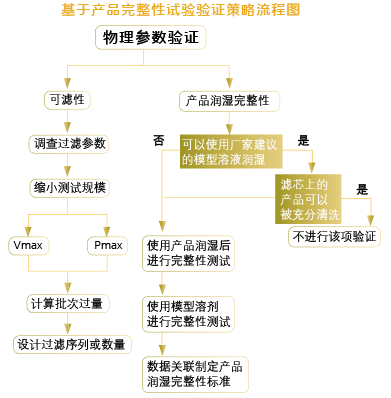
完成过滤工艺的验证之后,还应当定期评估产品性质和工艺条件,以确定是否需要进行再验证。
小结:
至少(但不限于)对以下内容进行评估,以决定是否需要开展再验证:
1. 单位面积的流速高于已验证的流速
2. 过滤压差超过被验证压差
3. 过滤时间超过被验证的时间
4. 过滤面积不变的情况下提高过滤量
5. 过滤温度变化
6. 产品处方改变
7. 过滤器灭菌条件或者灭菌方式改变
8. 过滤器生产商改变,过滤器生产工艺的变更,或者过滤器的膜材或结构性组成发生改变

 英文
英文  西语
西语 阿拉伯语
阿拉伯语 日语
日语